snRNA-seq
数据概览桑基图
大概是实验设计如此,并没有找到哪个数据包含了实验组条件对应的损伤模型、损伤后取样时间,在文中也没有显式提及,找了几个隐晦提及的地方:
- b, Susceptible and resilient subtypes of spinal cord neurons. Volcano plot shows log2 odds ratios comparing neuron proportions between the uninjured spinal cord and each injured condition at 7 days post injury (x axis) versus statistical significance (t-test, y axis).
- b,c, Chronophotography (b) and walking performance (n = 5 each) (c) of young and old mice after spontaneous recovery from moderate crush SCI.
实验设计大概就是:
- 一组未受伤组
- 损伤后时间组(1天、4天、7天、14天、1个月、2个月)和治疗组之类的全部为中等挤压损伤
- 不同类型损伤组(轻度挤压、中度挤压、严重挤压、完全挤压、挫伤、半切)和部分药物治疗组(甲强龙和米诺环素)全部为损伤后7天取样
- 其余为基因治疗组(ChABC和荧光蛋白的对照),为损伤后2月取样
然后手动写桑基图的数据,最后每个实验组的数量都一样,得出:
数据读取
主要是因为这个features.tsv的格式,Read10X()要用gene.column = 1参数。
质控
筛feature和count和线粒体基因与其他比起来有显著差异的低质量组,snRNA-seq筛掉了2个,应该先验证方差齐性之类的然后用ANOVA、kruskal检验之类的,但是跑了半天才发现读出来的数据已经是52只小鼠的435099个细胞了,所以跳过。
修正: 经过对比UMAP图,发现作者还是少了几团细胞,然而,数据中的最低nCount是201,所以作者给出的“原始数据”其实经过了质控,但是没有完全质控,现在发现少突胶质细胞的尾巴似乎可以通过DecontX来去除,另外一些可能要进行双细胞的筛选。
双细胞筛选
作者用了DoubletFinder和scDblFinder然后取并集。
对于DoubletFinder来说,似乎应该分组分得细一点,到library这个级别。
而对于scDblFinder来说,假如要用BiocParallel并行计算,似乎因为Matrix 1.6.5删除了一些函数,会报错,Matrix 1.6.1可以解决(但是最新的SeuratObject又需要Matrix 1.6.4以上,和nodeJS一样的麻)。
再次修正: 跑了一堆双细胞和DecontX之后,结果越来越奇怪,认为此数据已经经过完全质控,证据如下:
- 上述的原数据就已经是52只小鼠的435099个细胞,并且nCount最低是201
- 作者的元数据中有着5层的细胞类型数据
所以还是跳过。
整体可视化
PCA
作者说了用50维,但还是画个elbow图看一下效果,得到:
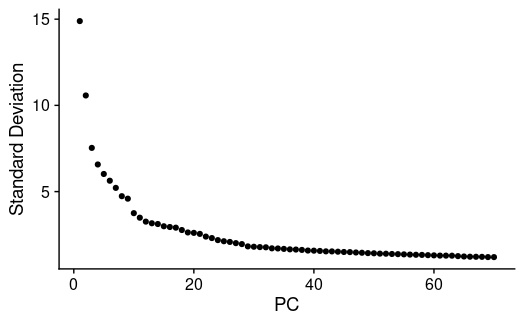
发现50维也还行(甚至有点过了)。
整合
reduction参数用'pca'会报错,但是默认参数就是PCA,不管。
Leiden法聚类
leiden法需要用到Python,R不搞代理和镜像,reticulate包可能安不上各种包,开root用pip安装leidenalg、numpy、igraph。
根据seurat#2294,对于这个435099个细胞的数据,leidenalg有一个很坑的地方就是会把数据转换成稠密矩阵,VIRT直接1400g,需要用method = "igraph"强制使用稀疏矩阵。
UMAP
用于可视化的降维,一个是原文用的UMAP,另一个是UMAP时间复杂度是min.dist参数设0.2比较好看点)
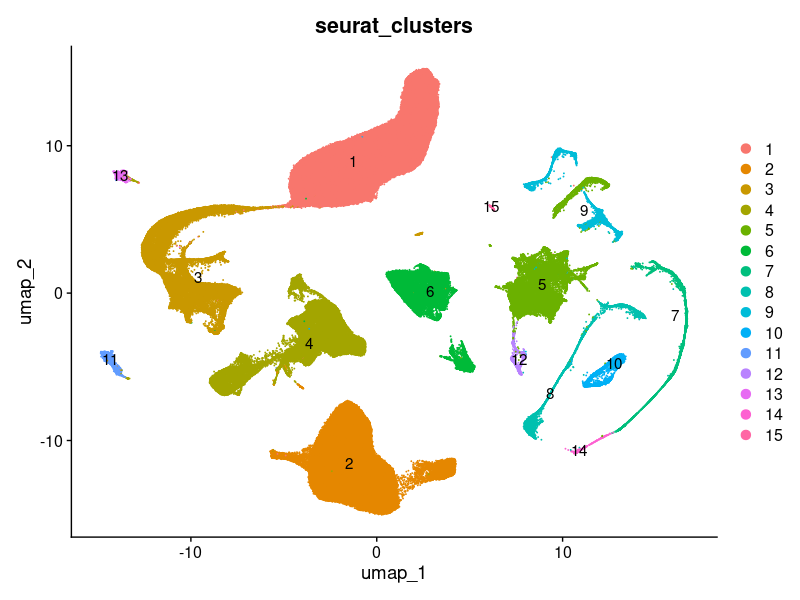
类型注释
说是manually annotated on the basis of marker gene expression, guided by previous studies,用几个marker试一下少突胶质细胞:
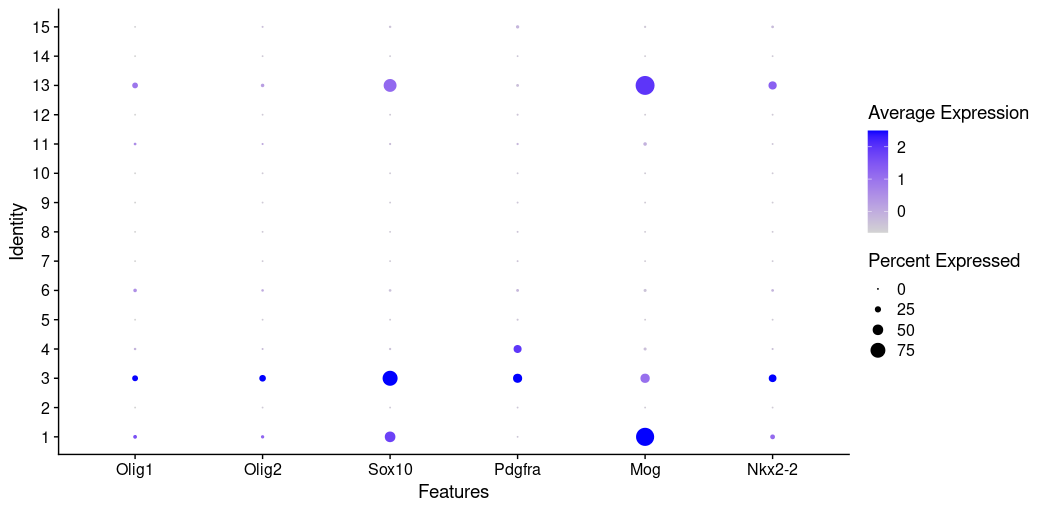
那标cluster 1、3、13为少突胶质细胞,和原文基本一致(但是原文13找不到)。
然后找了一些marker做了一张恐怖的图x
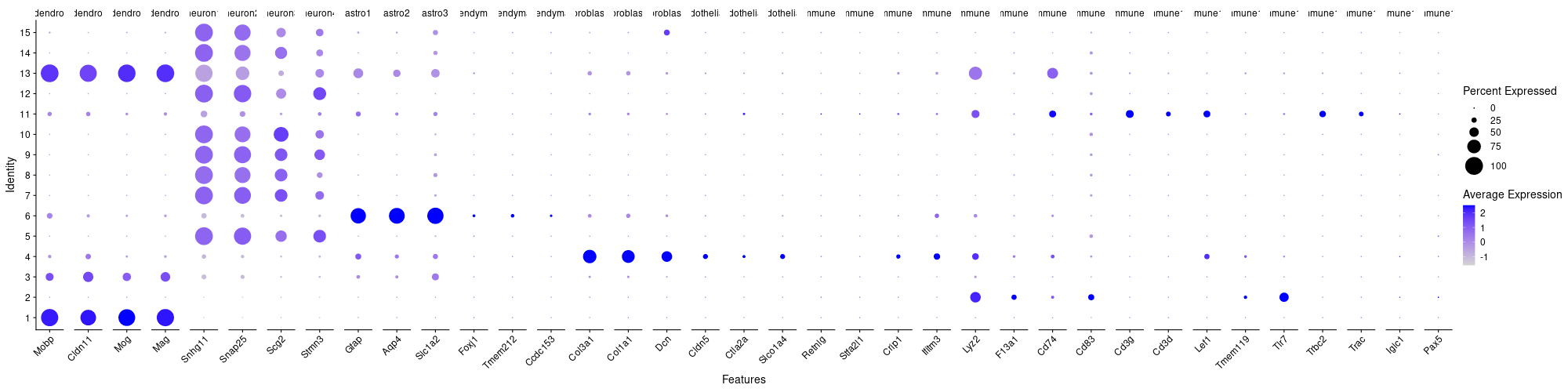
有一个报错是
1 | Error in `levels<-`(`*tmp*`, value = if (nl == nL) as.character(labels) else paste0(labels, : |
这是因为DotPlot()传入了两个重复的feature参数(在这里是marker)。
cluster
13感觉比较奇怪,似乎是少突胶质细胞和神经细胞的合体,试着用findDoubletClusters(),是阴性,而且听别人讲这个方法只能用于同批次的数据。
再次回顾元数据,将里面的cluster等字段用UMAP可视化,发现一些原文的cluster在经过上述分析后被分开了。
而作者声称在进行聚类之后删掉了一些cluster,结合上面质控时质控后二次分析结果越来越奇怪来看,我认为:
重复质控将去除某些cluster内过渡的液滴,使得原来的整个cluster被分割开
这个cluster 13或许也是有问题的,有可能是双细胞?但是先按着作者的思路不管,接下来仅走一个亚群注释的流程,在之后的分析使用作者的元数据。
可视化
先是原始数据的可视化,还没研究作者的图例:
然后是聚类结果的可视化: