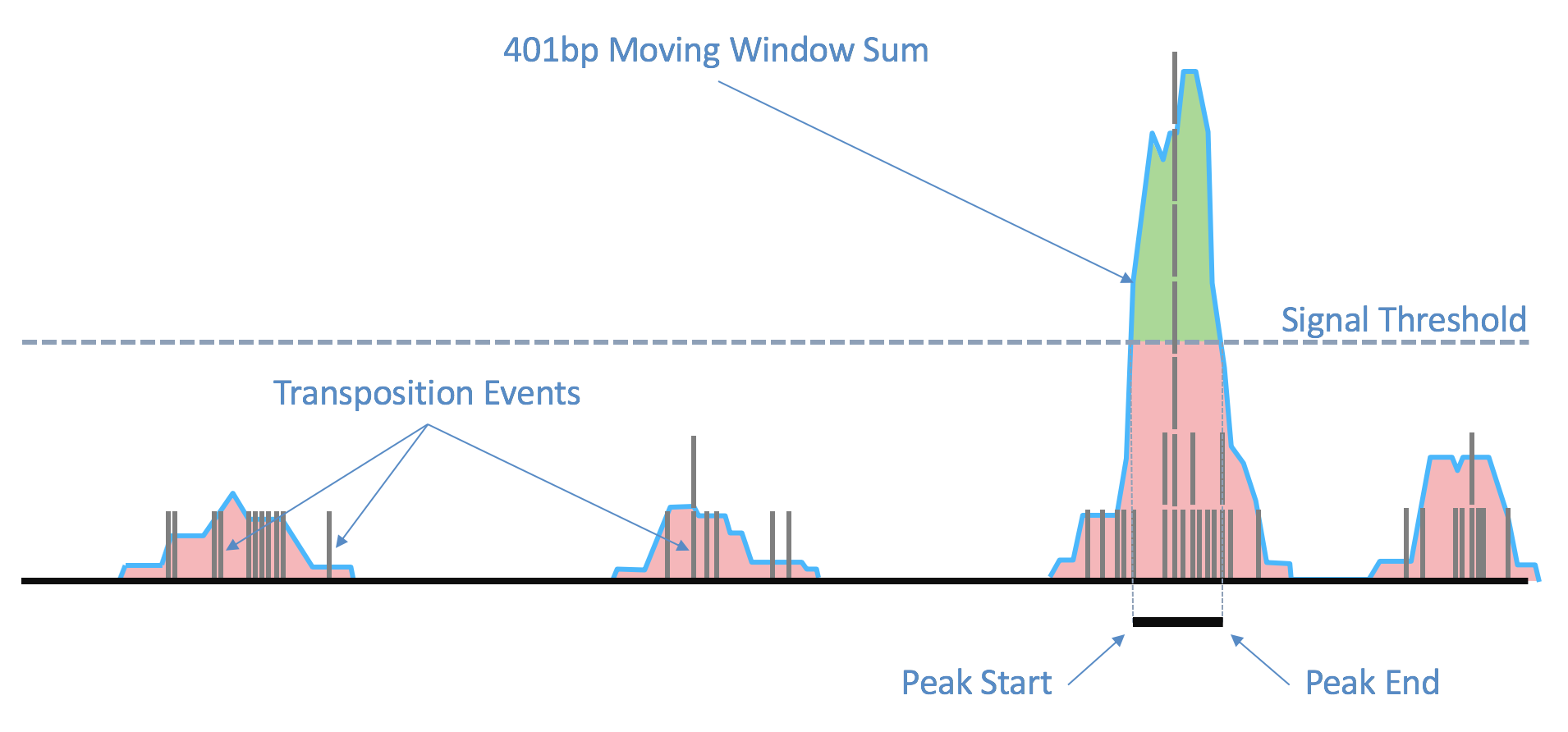
ATAC-seq的原理可以通过这个视频简单了解,在染色质开放区域,转座事件更容易发生,产生更多片段,形成peak。
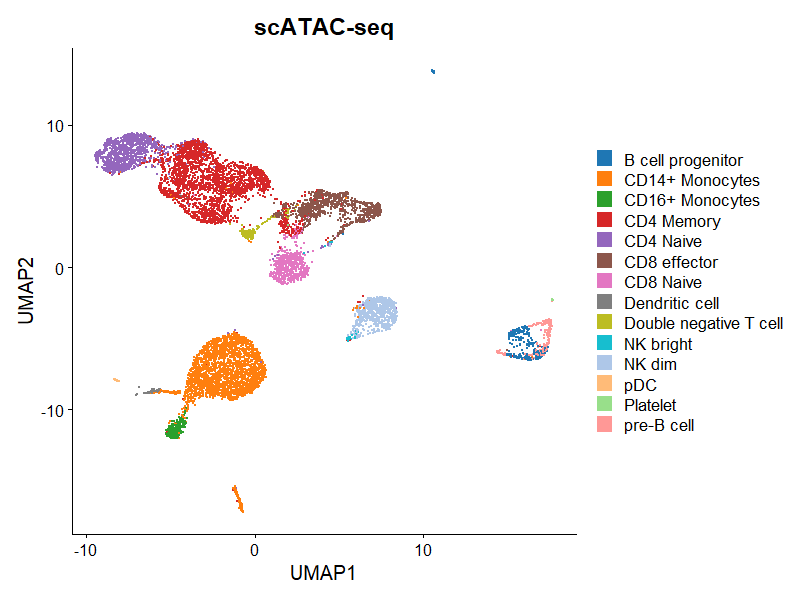
通过scATAC-seq分析,我们可以得出,例如,不同条件下染色质开放区域的差异,高度开放区域可能为活跃转录的基因。
这里使用Seurat V5和signac对PBMC进行scATAC-seq分析,原始代码见signac文档。
数据导入
(所以其实,可以只用片段信息来生成peak-细胞计数矩阵。)
还可以添加注释信息,便于下游分析直接从对象中读取。
生成Seurat对象
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
| pkgs <- c("Signac", "Seurat", "hdf5r", "GenomicRanges", "ggplot2", "patchwork")
pkgs_bioconductor <- c("AnnotationHub", "biovizBase")
for(pkg in pkgs){
if(!requireNamespace(pkg)) {
install.packages(pkg, dependencies = TRUE)
}
library(pkg, character.only = TRUE)
}
for(pkg in pkgs_bioconductor){
if(!requireNamespace(pkg)) {
BiocManager::install(pkg, update = FALSE, ask = FALSE)
}
library(pkg, character.only = TRUE)
}
counts <- Read10X_h5(filename = "10k_pbmc_ATACv2_nextgem_Chromium_Controller_filtered_peak_bc_matrix.h5")
metadata <- read.csv(
file = "10k_pbmc_ATACv2_nextgem_Chromium_Controller_singlecell.csv",
header = TRUE,
row.names = 1
)
chrom_assay <- CreateChromatinAssay(
counts = counts,
sep = c(":", "-"),
fragments = "10k_pbmc_ATACv2_nextgem_Chromium_Controller_fragments.tsv.gz",
min.cells = 10,
min.features = 200
)
pbmc <- CreateSeuratObject(
counts = chrom_assay,
assay = "peaks",
meta.data = metadata
)
ah <- AnnotationHub()
ensdb_v98 <- ah[["AH75011"]]
annotations <- GetGRangesFromEnsDb(ensdb = ensdb_v98)
seqlevels(annotations) <- paste0('chr', seqlevels(annotations))
genome(annotations) <- "hg38"
Annotation(pbmc) <- annotations
rm(list = setdiff(ls(), "pbmc"))
gc()
|
质控
- 有一些特征,peak,可能是染色体支架,或者不在22+X+Y染色体上,可以去除
- Nucleosome banding
pattern:通过将核小体相关片段和无核小体片段的比例量化为核小体信号,保留低信号(开放染色质区域占主导)的样本
- Transcriptional start site (TSS) enrichment
score:反映转录起始位点周围的片段数量(与背景的比值),一般TSS周围富集着开放区域之类的,低就是不好
- peak内片段数:低可能是因为测序深度不够,高可能是双细胞
- peak内片段数比例:低可能代表着低质量细胞或者伪迹
- 黑名单区域比例:一些区域内的片段代表着伪迹
质控
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
|
peaks.keep <- seqnames(granges(pbmc)) %in% standardChromosomes(granges(pbmc))
pbmc <- pbmc[as.vector(peaks.keep), ]
pbmc <- NucleosomeSignal(object = pbmc)
pbmc <- TSSEnrichment(object = pbmc)
pbmc$pct_reads_in_peaks <- pbmc$peak_region_fragments / pbmc$passed_filters * 100
pbmc$blacklist_ratio <- FractionCountsInRegion(
object = pbmc,
assay = 'peaks',
regions = blacklist_hg38_unified
)
pbmc <- subset(
x = pbmc,
subset = nCount_peaks > 9000 &
nCount_peaks < 100000 &
pct_reads_in_peaks > 40 &
blacklist_ratio < 0.01 &
nucleosome_signal < 4 &
TSS.enrichment > 4
)
|
线性降维、聚类和可视化
对于标准化,这里使用TF-IDF,其思想是,假如某特征,peak,在某样本,细胞,中频率较高(与Term
Frequency正相关),而包含其的其它细胞数较少(与Inverse Document
Frequency负相关),则这个peak的类别区分效果较好,得到的是Term
Frequency*Inverse Document Frequency。
对于选择用于降维的特征,由于scATAC-seq的低变化范围,可以选择总和最大的一些peak,或者丢弃仅在少数细胞中出现的peak。
对于线性降维,官方用SVD,然而PCA和SVD是可以互相转化的,而且PCA也多用SVD实现,就当成PCA了。
需要注意的是,对于scATAC-seq,第一个主成分常常与测序深度相关,可以检查每个主成分与测序深度的相关性,不使用相关性较高的。
然后一样的计算kNN图然后聚类,UMAP等降到2维用于可视化。
线性降维、聚类和可视化
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
| pbmc <- RunTFIDF(pbmc)
pbmc <- FindTopFeatures(pbmc, min.cutoff = 'q0')
pbmc <- RunSVD(pbmc)
depth_correlation <- DepthCor(pbmc, n = 50)
pcs_todrop <- c(which(lapply(depth_correlation$data$counts,abs) > 0.5))
pcs_filt <- setdiff(1:length(pbmc[["lsi"]]@stdev), pcs_todrop)
pcs_use <- pcs_filt[cumsum(pbmc[["lsi"]]@stdev[pcs_filt]^2) / sum(pbmc[["lsi"]]@stdev[pcs_filt]^2) < 0.9]
pbmc <- RunUMAP(object = pbmc, reduction = 'lsi', dims = pcs_use)
pbmc <- FindNeighbors(object = pbmc, reduction = 'lsi', dims = pcs_use)
pbmc <- FindClusters(object = pbmc, verbose = FALSE, algorithm = 3)
|
类型注释
通过与scRNA-seq整合进行类型注释,但是呢,需要相同的特征,基因,来寻找锚点,所以需要通过片段来预测ATAC-seq的基因活动性。
可以通过prediction.score.max检查准确性。
(这里因为血小板无核,不应该被ATAC-seq检测到,应该进一步探究,但它们很少,直接删掉。)
类型注释
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
|
gene.activities <- GeneActivity(pbmc)
pbmc[['RNA']] <- CreateAssayObject(counts = gene.activities)
pbmc <- NormalizeData(
object = pbmc,
assay = 'RNA',
normalization.method = 'LogNormalize',
scale.factor = median(pbmc$nCount_RNA)
)
DefaultAssay(pbmc) <- 'RNA'
pbmc_rna <- UpdateSeuratObject(pbmc_rna)
transfer.anchors <- FindTransferAnchors(
reference = pbmc_rna,
query = pbmc,
reduction = 'cca'
)
predicted.labels <- TransferData(
anchorset = transfer.anchors,
refdata = pbmc_rna$celltype,
weight.reduction = pbmc[['lsi']],
dims = pcs_use
)
pbmc <- AddMetaData(object = pbmc, metadata = predicted.labels)
predicted_id_counts <- table(pbmc$predicted.id)
major_predicted_ids <- names(predicted_id_counts[predicted_id_counts > 20])
pbmc <- pbmc[, pbmc$predicted.id %in% major_predicted_ids]
Idents(pbmc) <- pbmc$predicted.id
|
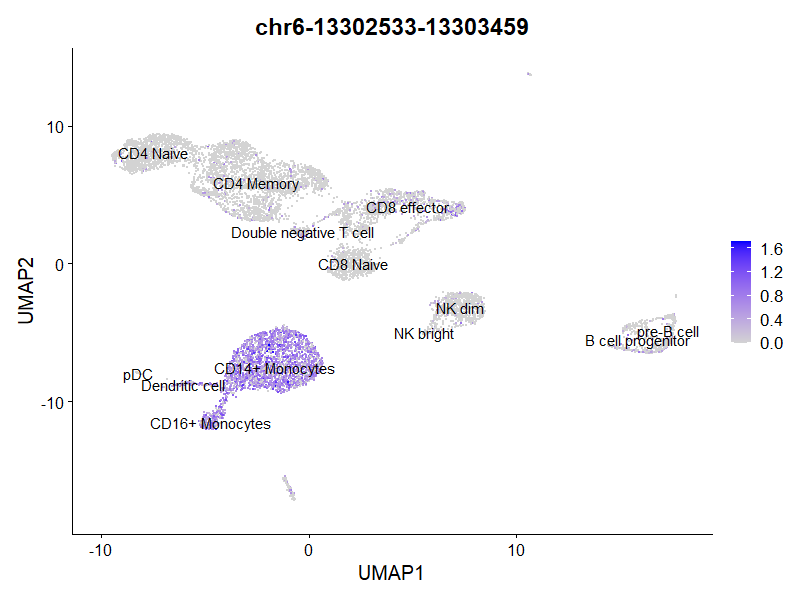
DA分析
类似地,我们可以在不同细胞类型间进行差异可及性分析,这里选CD4
Naive和CD14+ Monocytes。
但是只有peak有些难以解释,可以使用ClosestFeature()找到附近的基因,然后就可以做富集分析之类的了。
DA分析
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
| DefaultAssay(pbmc) <- 'peaks'
da_peaks <- FindMarkers(
object = pbmc,
ident.1 = "CD4 Naive",
ident.2 = "CD14+ Monocytes",
test.use = 'wilcox',
min.pct = 0.1
)
open_cd4naive <- rownames(da_peaks[da_peaks$avg_log2FC > 3, ])
open_cd14mono <- rownames(da_peaks[da_peaks$avg_log2FC < -3, ])
closest_genes_cd4naive <- ClosestFeature(pbmc, regions = open_cd4naive)
closest_genes_cd14mono <- ClosestFeature(pbmc, regions = open_cd14mono)
|
区域内Tn5插入频率可视化
最后,我们可以使用CoveragePlot()对不同簇、细胞类型等,可视化基因组感兴趣区域内的Tn5插入频率。
区域内Tn5插入频率可视化
1
2
3
4
5
6
7
8
9
10
11
12
13
14
|
pbmc <- SortIdents(pbmc)
regions_highlight <- subsetByOverlaps(StringToGRanges(open_cd4naive), LookupGeneCoords(pbmc, "CD4"))
CoveragePlot(
object = pbmc,
region = "CD4",
region.highlight = regions_highlight,
extend.upstream = 1000,
extend.downstream = 1000
)
|
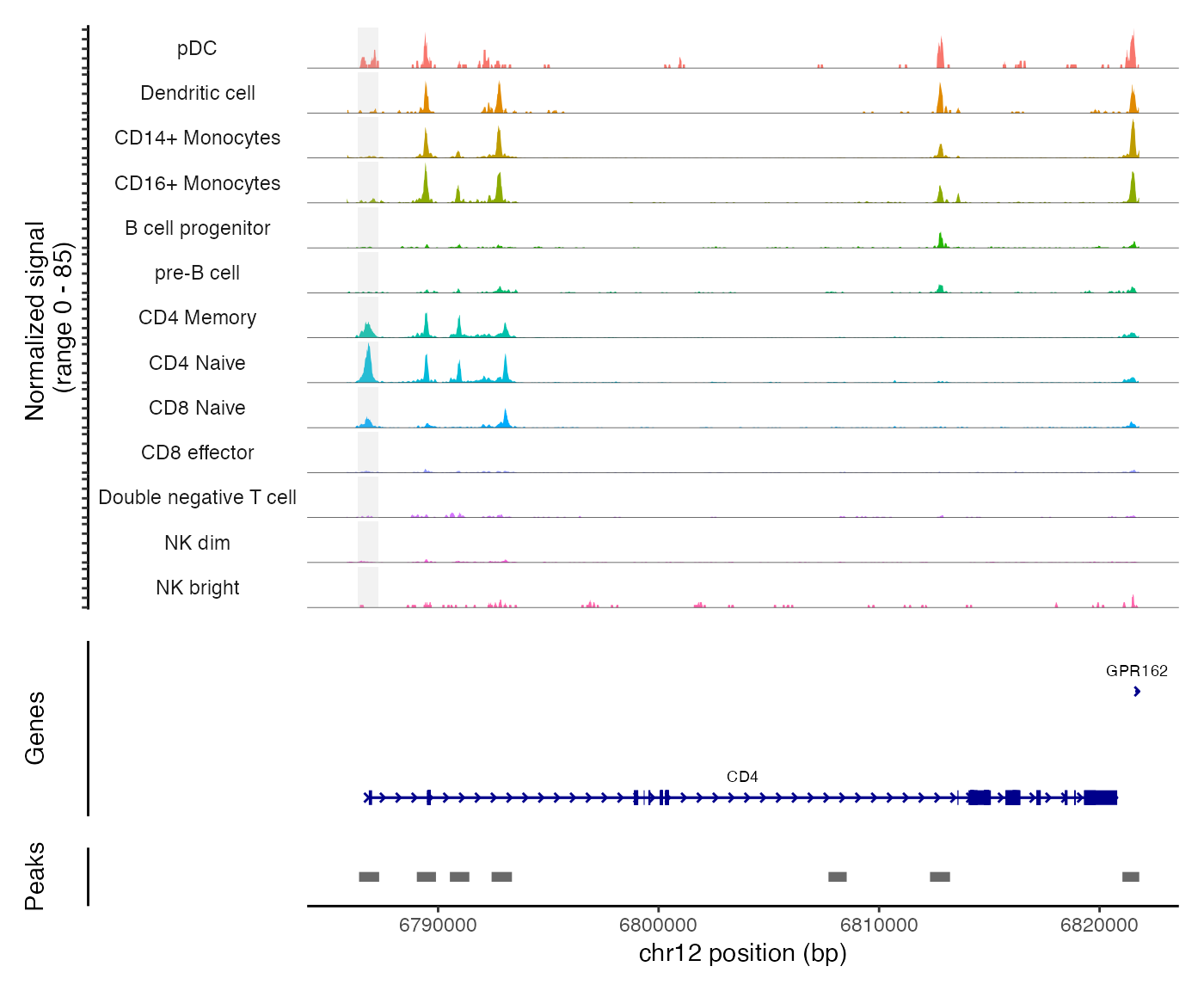
在这个图里,灰色那条带标记了差异可及peak。